Прогрессирующая мышечная дистрофия дюшенна
Содержание:
Миопатия: виды
В группу миопатий принято включать хронические, постепенно прогрессирующие заболевания, связанные с поражением, исчезновением мышечных волокон, заменой их жировыми или соединительными тканями. Четкой классификации на сегодня не существует.
Многие исследователи выделяют патологии по области преимущественного поражения, например, лице-лопаточно-плечевую, конечностно-поясную. Другие говорят о миопатиях в зависимости от характера причин – наследственных или приобретенных. Разделяют патологии по тому, что преимущественно поражается – белки или ферменты.
Принято также выделять отдельные болезни:
- Мышечная дистрофия Дюшенна. Болезнь встречается в 0,03% случаев. Развивается у мальчиков до 5 лет. Это одна из самых тяжелых форм миопатий. В среднем к 15 годам ребенок теряет возможность двигаться и самостоятельно себя обслуживать. Патология поражает сначала мышцы нижних конечностей, затем – верхних. В некоторых случаях развивается умственная отсталость.
- Ювенильная мышечная дистрофия Эрба-Рота. Заболевание регистрируется обычно в подростковом и юношеском возрасте до 20 лет. Поражаются мышцы таза и нижних конечностей. Больного отличает походка, напоминающая утку, тонкая талия. Отмечаются крыловидные лопатки. Обследование выявляет ухудшение сухожильных рефлексов. Из положения лежа больные встают с помощью рук.
- Плече-лопаточно-лицевая форма Ландузи-Дежерина. Миопатию этой формы отличает поражение мышц лица, плечевого пояса, лопаток. Страдают мышцы-разгибатели пальцев. Обнаруживается изменение формы груди. Заболевание впервые диагностируется в возрасте от 10 до 20 лет. В отличие от многих миопатий эта форма развивается медленно и имеет относительно благоприятный прогноз.
- Дистальная миопатия (тип Веландера). Появляется после 20 лет. Связана с уменьшением объема мышечной ткани в голеностопном отделе, коленях, предплечьях, кистях рук. Инвалидизация может наступить через 10 лет, а то и позже.
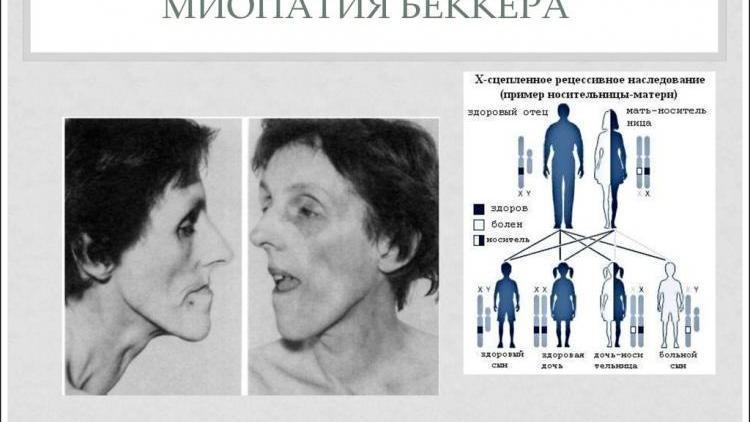
- Поздняя дистрофия Беккера. Обнаруживается у детей от 5 лет и молодых людей до 20 лет. Проявляется слабостью мышц, высокой утомляемостью, заменой мускульной ткани жировой. Сначала поражаются мышцы таза, бедер, голени, затем болезнь поражает руки. Интеллект остается сохранным.
Диагностика
Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
- На ЭКГ выявляется поражение миокарда латеральной и задне-нижней стенок левого желудочка, что определяется по следующим показателям: высокий зубец наблюдается в отведении V6; глубокий зубец Q наблюдается в отведениях V6, aVF, 2 и 3.
- Также исследуется содержание дистрофина в мышечной ткани (при этом заболевании дистрофия не выявляется).
- В ходе биохимических исследований в плазме крови определяется активность КФК (фермента креатинфорсфокиназы), которая обычно существенно повышена (в том числе у носительниц гена). Иногда для уточнения источника исследуются изоферменты КФК.
- Проводится также генодиагностика.
- Фибрилляции на ЭМГ сообщают о некрозе мышечных волокон.
- Биопсия мышц является одним из основных методов диагностики миопатии Дюшена, причем выбирается умеренно пораженная мышца, поскольку очень ослабленная и существенно пострадавшая мышца окажется неинформативной.
Наиболее достоверными являются анализы на активность в сыворотке мышечных ферментов, биопсия мышц и ЭМГ (электромиография).
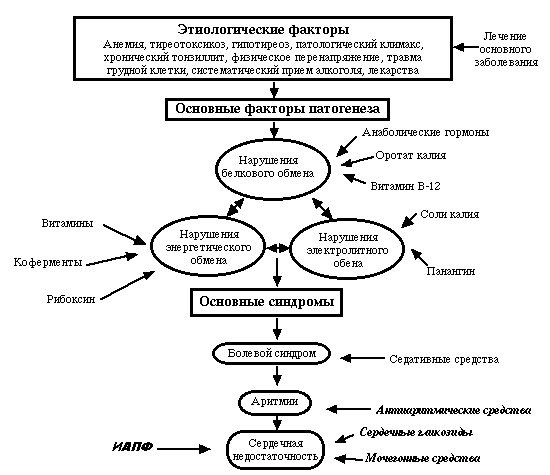
Кардиологическая помощь1, 19
- Симптомы сердечной недостаточности могут остаться незамеченными.20
- Необходимо провести ранний скрининг для выявления кардиомиопатии, прежде чем она станет симптоматической — например, шестимесячный обзор кардиологии с раннего детства.
- Стандартные тесты, такие как электрокардиограмма (ЭКГ) и эхокардиограмма (эхо), могут быть трудно интерпретировать из-за сколиоза.
- Сердечная недостаточность и аритмии лечатся стандартным способом с использованием ингибиторов ангиотензинпревращающего фермента (АПФ), диуретиков и бета-блокаторов.
- Лечить ночную гиповентиляцию (которая усугубляет проблемы с сердцем).
- Если даны глюкокортикоиды, необходим усиленный мониторинг сердечной деятельности и наблюдение за увеличением веса и артериального давления.
- Пациенты подвержены повышенному риску тромбоэмболии; Рассмотрим антикоагуляцию, если есть серьезные нарушения сердечной деятельности.
Процедуры и операции
Используются различные методы физиотерапии: парафиновые/грязевые аппликации, электростимуляция мышц, ультразвук, электрофорез (с нейромидином, неостигмином, прозерином, никотиновой кислотой), ионофорез с кальцием, легкий массаж, ЛФК. Особое значение отводится регулярно проводимой лечебной гимнастике, включающей пассивные и активные движения в различных положениях и во всех суставах: активные движения, рекомендуется выполнять в изометрическом режиме. Однако, необходимо предостеречь от чрезмерных нагрузок и особенно упражнений, сопровождающихся перерастяжением мышц
Немаловажное значение имеют дыхательные упражнения (особенно при иммобилизации больного)
По показаниям проводятся ортопедические операции по удлинению ахиллова сухожилия, исправление деформации стопы, рассечение фасций, пластика суставов.
Симптомы
Слабость и атрофия мышечных тканей считается главными признаками миопатии Эрба. Боли пациенты не испытывают, постоянная слабость не проходит после продолжительного отдыха. На первых стадиях заболевания возникают незначителные кратковременные улучшения после сна и отдыха. Спустя какое-то время слабость возобновляется.
Начинают проявляться крыловидные лопатки. Походка становится переливающейся. Грудь и живот выдвигаются вперед. Основной отличительной особенностью является гипомимическая физиономия с отчетливо выведенными вперед губами.
Преобразованию подлежит часть субнервального аппарата. Пациенты с миопатией Эрба отличаются тонкой талией. Быстро развиваются контрактуры, поэтому сокращаются мышечные ткани в области стоп и голеней. Пациент начинает хромать, опирается на большие пальцы. Концентрация жирных кислот в организме повышается. В кровеносной системе увеличивается уровень калия. Пациентам тяжело подниматься по ступенькам или вставать со стула.

При попытке подняться из лежачего положения пациент старается упереться руками и проделать несколько этапов для подъема. После этого мышечная дистрофия переходит в атрофию, мускулатура на руках и туловище истончается.
Процесс заболевания продолжительный, потому пациента, ощущая слабость, могут ходить самостоятельно до 45 лет и дольше.
Лечение
Возможности вылечить полностью заболевание в настоящее время нет. Терапия носит симптоматический характер, ее цель – продлить жизнь человека, сделать ее лучше. Используются лекарственные препараты, ортопедические средства, лечебная физкультура, физиопроцедуры, массаж. Рекомендовано регулярное проведение санаторно-курортного лечения.
Важным является разработка и внедрение новых способов лечения. Так, сегодня проводятся испытания стволовых клеток. По мнению ученых, они могут заменить поврежденные клетки мышц. Другим новым способом является генная терапия. Ее цель – активировать ген, который связан с выработкой белка утрофина. Ученые считают, что он аналогичен дистрофину и может восполнить его дефицит.
Лекарственная терапия
Уменьшить проявление симптомов миопатии помогает лечение глюкокортикостероидами (Преднизолон). Средства этой группы не имеют длительного эффекта и дают побочные реакции, например, приводят к лишнему весу, молочнице.
Улучшают передачу нервных импульсов ингибиторы ацетилхолинэстеразы, в частности, Прозерин. Лекарства бета-адреномиметики замедляют появление симптомов болезни, помогают повысить тонус и силу мышц. Улучшить обмен веществ в тканях помогут анаболические стероиды (Нандролон Деканоата).
Показано применение витаминов группы A, B, C, E.
Физиотерапия
Применение физиотерапевтических методов позволяет улучшить проводимость нервных импульсов к мышцам, улучшить их питание, обмен веществ, кровообращение.
Используют электрофорез, ультрафонофорез, бальнеотерапию, гидромассаж, лечение лазером.
Массаж
Основная цель массажа – повысить тонус мышц. Для получения эффекта его проводят несколько раз в год. Во многих случаях родственникам больного рекомендовано научиться приемам массажа для регулярного выполнения дома.
Физические упражнения
В терапии миопатии большое значение имеет лечебная физкультура. Комплекс элементов, их сложность зависят от степени развития патологии. В течение года проводится до 4 курсов ЛФК с физиотерапевтом в специальных центрах. В промежутках между курсами упражнения делают в домашних условиях. Отсутствие физической нагрузки ведет к быстрому развитию заболевания.
Больным рекомендовано посещать бассейн. Плавание, выполнение упражнений в воде способствует развитию мышц. При этом не возникает нагрузки на позвоночник. Помимо этого, нахождение в воде улучшает настроение, доставляет радость.

Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
 Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
https://youtube.com/watch?v=zctGZG1JgzY
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
 Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
 Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
https://youtube.com/watch?v=Rnjsh0bmHXA
Симптоматика
Новорожденные дети не имеют каких-либо явных отклонений в здоровье и строении. Первые клинические признаки появляются у малышей 1,5-2 лет. Болезнь прогрессирует, а симптоматика нарастает.
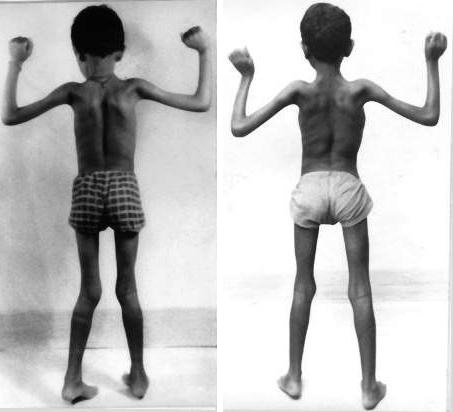
проявления миопатии Дюшенна
- Родители замечают, что их ребенок двигается неловко и неустойчиво, постоянно спотыкается и падает при ходьбе, не может прыгнуть, подняться по ступенькам, встать из лежачего или сидячего положения. Больные дети очень медлительны и неуклюжи во время двигательной активности. Это связано с поражением мышц таза и ягодиц, которые становятся слабыми первыми.
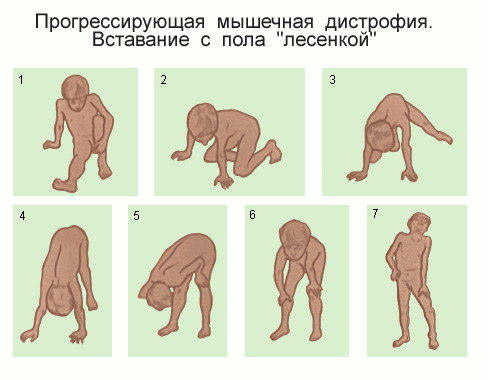
- Появляется характерный симптом Говерса: больные, поднимаясь с пола, помогают себе руками – они опираются на свои колени и бедра.
- Еще один патогномоничный признак миопатии Дюшена – ложная гипертрофия мышц голени, бедер, ягодиц: они кажутся большими, упругими, твердыми. На самом деле мышечная ткань разрушилась, а на ее месте разрослась фиброзная и жировая ткань.
- Больные дети испытывают страх перед ходьбой и с каждым днем становятся все более пассивными. Они с трудом стоят без помощи окружающих, быстро утомляются и отличаются низкой выносливостью. Мышечная слабость в ногах становится причиной формирования «утиной» походки — больные как бы переваливаются с ноги на ногу. При прогрессировании миопатии патологический процесс охватывает весь торс, в том числе и верхние конечности.
- У больных возникает поражение миокарда, протекающее в форме кардиомиопатии, причиной которой является также дефицит дистрофина. У детей возникают болезненные ощущения в груди, дыхание становится глубоким, неритмичным и частым. Даже на ранней стадии патологии появляются изменения на электрокардиограмме, характерные для коронарной недостаточности. Сердечно-сосудистые расстройства проявляются аритмией, лабильностью артериального давления, приглушенностью сердечных тонов.
- У части пациентов нарушается психоэмоциональное развитие, проявляющееся отставанием в обучении от ровесников, речевым расстройством в виде дислексии, плохой памятью. У 30% больных возникает олигофрения, аутизм. Интеллектуальные возможности у таких детей постепенно снижаются, порой достигая легкой степени идиотии. Невозможность посещать детские сады, школы и прочие общественные заведения еще больше усугубляет имеющиеся когнитивные расстройства.
- При прогрессировании патологии развиваются контрактуры крупных суставов, деформируется стопа, искривляется позвоночник по типу кифоза или лордоза, резко слабеют, а затем и полностью исчезают сухожильные коленные рефлексы.
- Миодистрофия Дюшенна всегда сопровождается повреждением грудины, которая приобретает неправильную форму и сильно изменяет тело человека. Из-за тяжелых атрофических процессов в мышцах талия становится «осиной», а лопатки «крыловидными».
- У 50% больных возникают эндокринные расстройства, проявляющиеся ожирением, гипоплазией и гипофункцией половых органов, низкорослостью.
Погибают больные обычно в возрасте 25 лет от неуклонно нарастающей слабости скелетных мышц, сформировавшейся стойкой дисфункции органов дыхания и сердечно-сосудистой системы.
Что такое миопатия Дюшена
Это серьезное заболевание поражает, главным образом, мышцы в области туловища, бедер и плеч. При этом пациенты, как правило, могут свободно использовать руки и пальцы, но у них возникают проблемы с ходьбой, бегом, и так далее. Мышечная слабость прогрессирует постепенно. Обычно она проявляется в раннем детстве, но поначалу симптомы миопатии Дюшена выражены очень слабо. С возрастом они становятся все более выраженными, и приводят к резкому снижению качества жизни.
Миопатия Дюшена диагностируется приблизительно у одного из 3500 мальчиков.
В мышечной ткани содержится дистрофин – белок, необходимый для нормальной работы мышц. У людей с миопатией Дюшена этого вещества слишком мало. Со временем это приводит к повреждению мышечных волокон и ослаблению мышц. Причиной этого является особый ген, который передается от родителей к детям, либо генные мутации, произошедшие в период внутриутробного развития.
Для каждого сына женщины, которая является носителем гена Дюшена, вероятность развития миопатии Дюшена составляет ровно 50%. Дочери такой женщины станут носителями этого гена с такой же вероятностью.
Если у ребенка миопатия Дюшена, значит ли это, что у кого-то из членов семьи есть ген Дюшена? Не обязательно. Приблизительно в половине случаев заболевшие миопатией этого типа не получают дефектный ген от одного из родителей. В клетках плода еще во время беременности происходят мутации, результатом которых и становится миопатия. Это может произойти из-за «ошибки», которая случилась при копировании родительских генов в клетки, которые должны будут образовать организм ребенка. Почему это происходит, в настоящее время неизвестно.
Точно узнать, есть ли у кого-то из членов вашей семьи ген Дюшена, можно, только при помощи генетического консультирования.
Каковы симптомы?
Обычно первые симптомы миопатии Дюшена появляются в возрасте 1-3 лет. Родители могут заметить следующие признаки миопатии Дюшена:
- Ребенку трудно ходить, бегать, прыгать, подниматься по лестницам. Походка ребенка может отличаться от походки его ровесников – он ходит вразвалочку, и менее уверенно, чем остальные. Иногда дети с миопатией Дюшена начинают ходить позже остальных, однако и совершенно здоровые малыши иногда делают первые шаги несколько позже сверстников;
- В более старшем возрасте ребенок может опираться на руки, чтобы встать;
- У ребенка могут наблюдаться проблемы с обучением – как правило, не очень серьезные.
Иногда первым признаком миопатии Дюшена является замедленное развитие речи.
Как диагностируется
В первую очередь врачи, как правило, просто наблюдают за ребенком, в особенности за тем, как он ходит, бегает и встает с пола. Если основания подозревать миопатию Дюшена, будет назначен анализ крови на креатинкиназу – это фермент, уровень которого у людей с этим нарушением всегда очень высок (в 10-100 раз выше нормы). Если уровень креатинкеназы у ребенка в норме, миопатию Дюшена исключают и начинают искать другие причины появившихся у малыша симптомов.
Следующим этапом диагностики миопатии Дюшена является биопсия мышечной ткани и/или генетическое тестирование.
В ходе биопсии врач берет небольшой фрагмент мышечной ткани для дальнейших анализов; процедуру проводят под общей анестезией. Образец ткани изучают под микроскопом при помощи особых техник, чтобы оценить состояние мышечных волокон и количество дистрофина.
Для проведения генетического тестирования необходимо необходимое количество крови пациента. С помощью этого метода выявляют гены, которые отвечают за развитие миопатии Дюшена. В большинстве случаев этот способ позволяет точно диагностировать данное заболевание.
Особенности каждой из форм болезни
Все формы болезни отличаются по локализации патологического процесса, типу наследования, возрасту начала проявлений. Также не все формы болезни встречаются с одинаковой частотой и являются в равной степени хорошо изученными.
Миодистрофия Дюшшена
Наиболее изученная форма патологии — миодистрофия Дюшшена. Эта форма имеет злокачественное течение и неблагоприятный прогноз. Как правило, в 14–15 лет больные уже полностью обездвижены. Ходить самостоятельно ребенок не может уже в 8–10 лет.
Начинается патологический процесс с ног и пояса нижних конечностей. Распространение происходит повосходящей. После нижних конечностей в него вовлекаются мышцы спины, рук, плечевого пояса. На термальной стадии развития затрагиваются мышцы глотки, лица, дыхательные.
 К первым признакам относится нарушение походки и псевдогипертрофия — визуальное увеличение и уплотнение мышц
К первым признакам относится нарушение походки и псевдогипертрофия — визуальное увеличение и уплотнение мышц
Первыми затрагиваются икроножные мышцы, но возможно проявление псевдогипертрофии и в других областях:
- ягодицы;
- дельтовидные мышцы;
- пресс;
- язык.
Сердечная мышца страдает достаточно часто, причем развиваются нарушения на ранних стадиях патологического процесса. Больные дети часто страдают от умственной отсталости. В разных случаях степень проявления олигофрении разная, предполагается, что зависит это от наследственных особенностей.
Миодистрофия по Беккеру
Схожая по клиническим проявлениям с миодистрофией Дюшшена, эта форма болезни отличается доброкачественным течением. При наследовании часто наблюдается так называемый эффект деда. Так называют случаи, когда больной передает внуку через дочь патологический ген. Такой вариант возможен из-за того, что больные дольше сохраняют трудоспособность и их фертильность не страдает, как среди больных миодистрофией Дюшшена.
Первые проявление болезни начинаются в 10–15 лет. Часто до 30 лет больной еще способен ходить — иногда и дольше. При этом интеллект больных не страдает, то есть олигофрения не наблюдается. Также кардиомиопатия развивается только в редких случаях.
Редкие формы болезни
К наиболее редким формам болезни, характеризующимся более мягким течением, относятся:
Рекомендуем вам почитать:Что такое миастения гравис
- миодистрофия Дрейфуса-Когана;
- форма Мэбри;
- миодистрофия Роттауфа-Мортье-Бейера.
Первая форма болезни отличается от остальных тем, что у больных с ней не развивается псевдогипертрофия мышц. Также умственные способности у человека сохранены, а кардиомиопатия начинает развиваться после 30–40 лет.
Форма Мэбри не имеет характерных для X-хромосомных патологий маркеров, хоть и передается по этой хромосоме. Сильно выражена псевдогипертрофированность мышц.
Для формы Роттауфа-Мортье-Бейера характерно нарушение сгибательных способностей во многих суставах. Начинается этот процесс с дистальных отделов ног, потом затрагивается шея, постепенно процесс переходит на весь позвоночник. У больного формируется постоянное патологическое положение головы из-за нарушений сгибания шеи.
 У больных развиваются парезы, но выражаются они умеренно: чаще всего затрагивается плечевой пояс
У больных развиваются парезы, но выражаются они умеренно: чаще всего затрагивается плечевой пояс
Болезнь очень медленно прогрессирует, поэтому многие больные сохраняют полную трудоспособность практически на протяжении всей жизни. Наиболее вероятная причина летального исхода — кардиомиопатия. Смерть наступает, чаще всего, в возрасте 40–50 лет.
Ювенильная миопатия Эрба
Первые симптомы болезни появляются достаточно поздно, но известны случаи псевдодюшшеновской миодистрофии Эрба. В этом случае первые симптомы развиваются в возрасте до 10 лет. Течение болезни более тяжелое, чем у тех больных, у которых первые проявления были обнаружены позже. Интеллектуальные способности у больных обычно сохранены. Патологический процесс начинается обычно с тазового пояса, затем затрагивает плечевой. В некоторых случаях они страдают одновременно.
Лице-лопаточно-плечевая форма
Миодистрофия Ландузи-Дежерина чаще встречается у женщин. Эта форма характеризуется сравнительно простым течением, но усугубить его могут чрезмерные физические нагрузки, в том числе нерациональная лечебная физкультура.
Чаще всего больные живут долго — до 60 лет и даже дольше. Патологический процесс распространяется с лицевых мышц на плечевой пояс, а затем — на проксимальные отделы рук. После этого иногда возможно распространение патологии на нижние конечности. Часто мышцы затрагиваются асимметрично.
Этиология
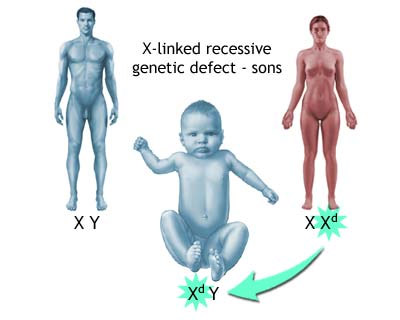 Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Мутантный ген, обуславливающий развитие миопатии, получил название ген Дюшенна. Он отвечает за синтез белка дистрофина, который обеспечивает целостность мышечных волокон во время сокращения. Белок составляет основу миофибрилл, поддерживает их клеточный скелет, позволяет им активно и многократно сокращаться и расслабляться. У лиц с миопатией он отсутствует или вырабатывается в очень незначительном количестве. Мышечные волокна постепенно разрушаются и замещаются фиброзной тканью или жиром. В результате серьезно страдает функция движения у больных.