Фенилкетонурия
Содержание:
Последствия и прогноз жизни
Воздействие излишнего количества фенилаланина на нервную систему ребенка приводят к стойким психологическим нарушениям. Уже к 4 годам, без должного лечения, больные фенилкетонурией дети причисляются к слабоумным и физически недоразвитым членам общества. Они пополняют ряды инвалидов детства и краски жизни меркнут для них.
Не искрится счастьем и жизнь родителей больного ребенка. Малыш требует постоянного ухода, и при ограниченных финансовых средствах это выливается в общее ухудшение благосостояния семьи. Боль, испытываемая мамой и папой от невозможности изменить существование ребенка в лучшую сторону угнетает и давит, но отчаиваться нельзя. Помогите себе, помогите ребенку пройти эти испытания с меньшими потерями в любви и милосердии.
Наука спешит, она делает семимильные шаги в направлении исключения заболевания из ранга тяжелых. Огромное значение имеет диагностика фенилкетонурии в утробе матери, но пока такого метода не изобретено. «пока» не значит «никогда», будем ждать и верить
Лечение
Болезнь фенилкетонурия в России лечится посредством диетотерапии, ограничивающей употребление белковых продуктов. В мире ведутся разработки препаратов, которые позволят контролировать уровень фенилаланина без соблюдения жесткой диеты. Перспективными направлениями исследований считаются:
- применение растительного фермента фенилаланинлиазы;
- генотерапия, направленная на коррекцию мутировавшего гена, вызывающего нарушения аминокислотного обмена;
- попытки введения гена фенилаланингидроксилазы в пораженные клетки печени.
Постоянное наблюдение у врачей (педиатра, невролога), соблюдение диеты, ограничивающей употребление белка, от рождения до половой зрелости является главным условием нормального развития ребенка, больного классической формой фенилкетонурии. Контроль уровня фенилаланина проводят регулярно: в первые 3 месяца жизни – один раз за неделю, от 3-х месяцев до года – минимум раз за месяц, от года до 3-х лет – 1 раз за два месяца, затем – по рекомендациям врача (около 4 раз за год). Количество употребляемых больным белков корректируется в зависимости от возраста и нагрузок.
Диетотерапия
Диета при фенилкетонурии основана на исключении из рациона больного животных белков. При ее постоянном строгом соблюдении удается предотвратить развитие церебральных нарушений, нарушений функции печени, если заболевание диагностировано в первые недели жизни. К строго запрещенным продуктам относятся:
- все виды мясных продуктов, колбасных изделий;
- все виды рыбы, морепродуктов;
- сыр;
- творог;
- яйца;
- орехи;
- хлебобулочные, кондитерские изделия;
- соевые продукты;
- крупы, хлопья.
Ежедневный рацион больного должен содержать овощи, фрукты, ягоды и зелень; разрешено употребление меда и сахара, крахмала, рисовой и кукурузной муки, сливочного, растительного масла. Ряд продуктов необходимо частично ограничить. С разрешения лечащего врача возможно употребление небольшого количества:
- молочных продуктов;
- картофеля, белокочанной капусты;
- овощных консервов.
Необходимые для развития и роста ребенка аминокислоты содержат специальные лечебные смеси, очищенные от лактозы, содержащие пептиды (молочные белки, уже расщепленные ферментами) и свободные аминокислоты (триптофан, тирозин, таурин, цистин, гистидин). Они производятся в форме порошков, которые разводят водой или сцеженным грудным молоком (при условии соблюдения матерью специальной диеты). Такими смесями и специальными продуктами для детей разного возраста являются:
- Афенилак;
- Мдмил-ФКУ-0;
- Аналог-СП;
- Апонти;
- Берлафен;
- Минафен;
- Лофенилак;
- Нофелан;
- Тетрафен;
- Циморган.
Медикаментозная терапия
Даже при регулярном получении белковых гидролизатов и аминокислотных смесей, больные фенилкетонурией нуждаются в дополнительном назначении комплексов витаминов (например, группы В) и минеральных соединений (содержащих препараты железа, фосфор и другие микроэлементы). Проводят симптоматическое и патогенетическое медикаментозное лечение с применением:
- препаратов карнитина для профилактики его дефицита (Элькар, L-карнитин);
- антиконвульсанты при судорожных припадках (Депакин, Клоназепам);
- ноотропные препараты для поддержания высших психических функций головного мозга (Пирацетам);
- средства, стимулирующие сосудистую микроциркуляцию;
- при атипичных формах фенилкетонурии, не поддающихся диетотерапии – гепатопротекторы, препараты с Леводопой, 5-окситриптофан, Тетрагидробиоптерин.
Широко применяется лечебная гимнастика, общий массаж. Реабилитация детей с фенилкетонурией предусматривает специальные методы педагогической подготовки во время дошкольного и школьного обучения. Больные нуждаются в регулярной помощи логопеда, педагога, дефектолога. Пациентам свойственна повышенная утомляемость, иногда требуется коррекция поведения, поэтому такие дети требуют к себе повышенного внимания и особого подхода.
Фенилкетонурия – что это за болезнь?
Фенилкетонурия, или болезнь Феллинга, является тяжелой патологией, впервые описанной в 1934 году норвежским ученым Феллингом. Тогда Феллинг провел обследование нескольких детей с умственной отсталостью и выявил у них присутствие в моче фенилпирувата – продукта распада поступающей с едой аминокислоты фениланина, которая в организме больных не расщепляется. Фенилкетонурия – заболевание, связанное с нарушением обмена веществ врожденного характера, открытое одним из первых.
Фенилкетонурия – тип наследования
Болезнь Феллинга является хромосомно-генетической, наследственной, передаваемой детям от родителей. Виновником развития патологии выступает ген, находящийся на 12 хромосоме. Он ответственен за производство печеночного фермента фенилаланин-4-гидроксилазы, за счет которого происходит превращение фенилаланина в другое вещество – тирозин (оно требуется для нормальной работы организма).
Установлено, что фенилкетонурия наследуется как рецессивный признак. Приблизительно 2 % людей являются носителями дефектного гена, но при этом не страдают фенилкетонурией. Патология развивает только тогда, когда и мать, и отец передают ген ребенку, а случиться это может с вероятностью 25 %. Если фенилкетонурия наследуется как рецессивный признак, жена гетерозиготна, а муж гомозиготен по нормальному аллелю гена, то вероятность того, что дети будут здоровыми носителями гена фенилкетонурии, равна 50 %.
Формы фенилкетонурии
Рассматривая, у кого может развиться фенилкетонурия, что это за заболевание, зачастую речь ведется о классической форме патологии, которая встречается примерно в 98 % случаев. Остальные случаи – кофакторная фенилкетонурия, обусловленная дефектом тетрагидробиоптерина вследствие нарушения его синтеза или восстановления активной формы. Данное вещество служит кофактором ряда ферментов, и без него невозможно проявление их активности.
Фенилкетонурия – причины
Болезнь Феллинга является патологией, при которой из-за мутаций в гене, вызывающих дефицит или отсутствие фенилаланин-4-гидроксилазы, происходит накопление в тканях и физиологических жидкостях фенилаланина, а также продуктов его неполного расщепления. Часть избыточного фенилаланина превращается в фенилкетоны, выводимые с мочой, что и обусловило название болезни.
Нарушение обменных процессов сказывается в большей степени на головном мозге. На его ткани производится отравляющее воздействие, нарушаются процессы жирового обмена, происходит сбой миелинизации нервных волокон, снижение образования нейромедиаторов. Так начинается запуск патогенетических механизмов задержки умственного развития у ребенка.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин
Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Лечение фенилкетонурии
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина.
Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
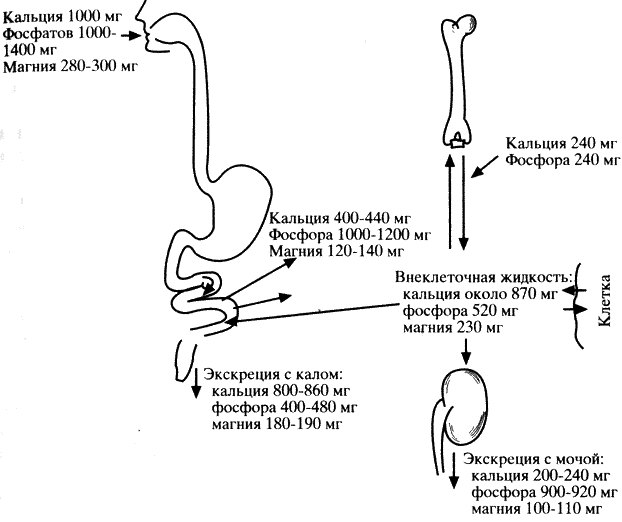
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.
Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии. В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций. Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину. Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина. В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения.
Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.
В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Данная статья опирается на статью из книги «Врожденные и наследственные заболевания» под редакцией профессора П.В.Новикова, М., 2007
Диагностика
Медико-генетическое консультирование семьи
Будущим родителям нужно быть особенно внимательными, если в семьях кого-либо из них имели место случаи рождения больных фенилкетонурией детей. Нельзя исключить возможность появления на свет ребёнка с ФКУ, даже если недуг проявился у дальнего родственника. Коварство заболевания кроется в бессимптомном носительстве повреждённого гена абсолютно здоровыми людьми.
Всем парам, у которых имеются подозрения на наличие наследственных болезней в роду, стоит обратиться к врачу-генетику ещё во время планирования малыша. В медико-генетическом центре (МГЦ) с помощью современных методов диагностики можно выявить носительство мутантного гена и рассчитать риск рождения ребёнка с ФКУ и другими генетическими болезнями.
Инвазивные методы диагностики (хорионбиопсии, амниоцентеза, кордоцентеза) могут помочь определить недуг до рождения крохи. При этом исследуется генетический материал, полученный от плода. Данные методы являются травматичными и могут повлечь за собой спонтанное прерывание беременности. Поэтому применение их оправдано лишь при доказанном носительстве мутантных генов у родителей и высоком риске возникновения ФКУ у малыша.
Неонатальный скрининг

Перед выпиской из родильного дома новорождённых малышей массово обследуют на наследственные заболевания
Для этого важного исследования медработник набирает кровь из пяточки малыша и наносит капельки биологической жидкости на фильтровальную часть тест-бланка. Каждая капля крови предназначена для выявления одного из 5 заболеваний: фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии
Неонатальный скрининг – очень важное и необходимое всем детям без исключения обследование. Он проводится совершенно бесплатно и направлен на сохранение здоровья нации
Отказываясь от манипуляции, родители совершают огромную ошибку, ведь симптомы наследственных недугов не всегда заметны у новорождённого малыша. Яркие клинические проявления нередко возникают, когда помочь ребёнку уже сложно, а изменения в организме крохи приняли необратимый характер.
Этот метод является достоверным в выявлении фенилкетонурии, если малыш получает достаточное количество энтерального питания, грудного молока. Поэтому новорождённым, которые находятся в отделении реанимации, анализ проводят позже. Отличаются и сроки постановки пробы у доношенных и недоношенных детей. Малышам, которые появились в срок, рекомендован набор капиллярной крови на 4-е сутки жизни. Проведение исследование родившимся преждевременно крохам откладывается до 7-х суток.
Анализ следует проводить натощак, что обеспечит более точный результат и повысит диагностическую значимость теста. Бланк с капельками крови малыша и паспортными данными родителей отправляется в лабораторию медико-генетической консультации, где проводится биохимическое исследование.
Родителям нужно обратить внимание на правильность заполнения бланка, корректность своих контактных данных. На проведение исследования уходит в среднем 10 дней, семья к этому времени обычно находится дома
В случае обнаружения положительного результата теста, родителям нужно будет в кратчайшие сроки обратиться в медико-генетический центр, а неправильные контактные данные задержат дальнейшее обследование малыша.
Обследование ребёнка в МГЦ
Чтобы подтвердить заболевание у младенца и установить его причину, малышу придётся пройти многоэтапную процедуру обследования. Повторные биохимические анализы помогут подтвердить повышение уровня аминокислоты в крови и принять решение о необходимости диетотерапии. Проводится исследование уровней фенилаланина и тирозина, активность печёночных ферментов, определение продуктов обмена аминокислот в моче.
Затем осуществляется молекулярно-генетическая диагностика, которая позволяет выявить мутацию в гене РАН, ответственном за развитие фенилкетонурии. С помощью данных методов можно определить и бессимптомное носительство заболевания.
Как развивается фенилкетонурия
Врожденное наследственное заболевание, вызываемое нарушением обмена аминокислот, называется фенилкетонурией. Другие названия этой патологии – болезнь Феллинга (по имени впервые описавшего болезнь врача) или фенилпировиноградная олигофрения (основными последствиями прогрессирования заболевания являются тяжелые поражения центральной нервной системы и головного мозга, провоцирующие задержки развития, умственную отсталость).
В России фенилкетонурия регистрируется с частотой 1 случай на каждые 5-10 тыс. новорожденных, в Турции эта пропорция составляет 1:2500, в Японии и некоторых странах Европы – 1:100 000, в странах Африки заболевание практически не встречается. По статистике девочки наследуют патологию в два раза чаще, чем мальчики. В неонатальном периоде клинические проявления болезни отсутствуют, но из-за поступления фенилаланина в организм с пищей манифестация фенилкетонурии происходит в первые полгода жизни.
При болезни Феллинга дефицит печеночного фермента фенилаланин-4-гидроксилазы вызывает нарушения метаболизма фенилаланина, поступающего с белковой пищей. Происходят следующие процессы:
- Запускаются побочные пути окисления фенилаланина, при которых в организме накапливаются его токсичные производные – фенилуксусная, фенилмолочная и фенилпировиноградная кислоты.
- Возникает дефицит тирозина (аминокислота, продукт нормального метаболизма фенилаланина), участвующего в функциях гипофиза, надпочечниках и щитовидной железы, выработке меланина, регулирующего аппетит и процессы жировых отложений.
- Развиваются нарушения обмена липопротеидов, гликопротеидов, белков.
- Происходит расстройство транспорта аминокислот.
- Нарушается обмен катехоламинов и серотонина.
- Образуется избыток фениэтиламина и ортофенилацетата, провоцирующий нарушения липидного метаболизма в головном мозге.
- Разрушение механизма передачи импульсов между клетками нервной системы из-за возникающего дефицита нейромедиаторов.
Причины
Синдром фенилкетонурия у детей в 98% случаев развивается вследствие мутации расположенного на длинном плече 12 хромосомы гена, кодирующего количество фенилаланингидроксилазы (что приводит к недостаточности этого фермента). Генетическая патология носит аутосомно-рецессивный характер наследования, то есть для развития болезни новорожденный должен унаследовать копию дефектного мутировавшего гена и от отца, и от матери, являющихся его гетерозиготным носителем. Этим обусловлена низкая степень распространенности патологии.
Формы и типы
Классическая фенилкетонурия 1 типа, развивающаяся в результате наследования от обоих родителей мутировавшего гена, составляет 98-99% от всех зарегистрированных случаев. Другими, атипичными формами болезни, не поддающимися лечению диетотерапией, но протекающими с аналогичными клиническими проявлениями, являются:
- фенилкетонурия 2 типа, развивающаяся в результате дефицита дегидроптеринредуктазы;
- фенилкетонурия 3 типа, возникающая из-за недостаточности тетрагидробиоптерина.
Online-консультации врачей
| Консультация гастроэнтеролога детского |
| Консультация проктолога |
| Консультация сексолога |
| Консультация диагноста (лабораторная, радиологическая, клиническая диагностика) |
| Консультация педиатра |
| Консультация иммунолога |
| Консультация детского психолога |
| Консультация гастроэнтеролога |
| Консультация оториноларинголога |
| Консультация маммолога |
| Консультация педиатра-аллерголога |
| Консультация сурдолога (аудиолога) |
| Консультация вертебролога |
| Консультация офтальмолога (окулиста) |
| Консультация психоневролога |
Новости медицины
Digital Pharma Day. Будьте во главе digital-трансформации фармацевтической индустрии,
09.10.2020
В сети EpiLaser самые низкие цены на ЭЛОС эпиляцию в Киеве,
14.09.2020
Щедрость продлевает жизнь,
07.09.2020
Универсальной диеты не существует, — ученые,
19.06.2020
Новости здравоохранения
Скорость распространения COVID-19 зависит от климатических условий,
11.06.2020
Исследователи насчитали шесть разновидностей коронавируса,
11.06.2020
Ученые выяснили, за какое время солнце убивает COVID-19,
28.05.2020
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Симптомы

С момента рождения ребенок выглядит абсолютно здоровым, поэтому важно с первых дней жизни выявить недуг. Первые симптомы фенилкетонурии возникают у ребенка в первом полугодии
При поступлении в организм малыша грудного молока или его заменителей развиваются начальные признаки: вялость, беспокойство, ребенок мало двигается, не исключено появление судорог, упорной рвоты, возможна гипервозбудимость. Слабо реагируя на происходящее, ребенок не узнает мать. По достижении шести месяцев становится заметным отставанием в психическом развитии.
У детей с такой патологией поздно появляются зубы, сидеть и ходить они начинают гораздо позже своих сверстников. В год малыши не понимают речь родителей, не способны выразить эмоции голосом. Фенилаланин выводится из организма с мочой и потом, поэтому от пациента исходит специфический затхлый запах, напоминающий мышиный.
Позже появляется характерное положение тела: ноги широко расставлены, согнуты в коленных и тазобедренных суставах, плечи и голова опущены. Походка мелкими шагами, с небольшим покачиванием. Из-за повышенного мышечного тонуса сидит такой пациент в позе портного — поджав ноги. Отмечается недостаток пигмента, у больного малыша светлые глаза и гипопигментированные волосы. Нередко возникает экзема, дерматиты, катаракта. К четырем годам у такого ребенка речь полностью отсутствует. Без проводимого лечения возникает микроцефалия, тремор рук, шаткая походка, акроцианоз (синюшность кожи), прогнатия (аномалия прикуса), гиперкинезы (непроизвольные движения). Также возможна дисфункция печени.
Диагностика фенилкетонурии
На сегодняшний день все новорожденные дети массово обследуются на наличие фенилкетонурии. На территории России этот вопрос регламентирует приказ Минздрава РФ №316 от 30.12.1993 г. Процедура получила название неонатальный скрининг и является эффективным способом выявления наиболее распространенных наследственных заболеваний, среди них и ФКУ.
Массовое обследование новорождённых это простой и достоверный метод диагностики. В роддоме у каждого ребенка берут несколько капель периферической крови из пяточки. Это делается натощак, через три часа после кормления. У доношенных детей анализ берут на четвертый день жизни, а у недоношенных на седьмой
У тех новорожденных, которые появились на свет не в родильных домах, важно взять анализ на протяжении первых трех недель
Кровь наносят на специальный тест-бланк, который потом отправляют в лабораторию для проведения генетического исследования. Там на протяжении суток проводится анализ крови на содержание в ней аминокислоты — фенилаланина. Результаты теста заносятся в обменную карту ребенка в виде штампа: «На ФКУ и ВГ обследован».
В том случае, если в анализе обнаруживают измененный ген, то родителей с ребенком приглашают в медико-генетический центр для обследования. Для того, чтобы подтвердить или опровергнуть диагноз назначаются дополнительные исследования:
- в сухом пятне крови
- в сыворотке крови
- потовый тест
- копрограмма
- ДНК-диагностика