Синдром клайнфельтера: мужчина с женской хромосомой
Содержание:
- Генетические и хромосомные заболевания плода
- Причины мутаций хромосом
- Лечение[править | править код]
- Профилактика генетических патологий плода
- Можно ли вылечить или предотвратить хромосомные аномалии
- Хромосомные аномалии плода. Признаки
- Виды хромосомных мутаций
- 2.Синдром Патау
- Анализ на кариотип супругов
- Синдром кошачьего крика — хромосомная аномалия у детей
- Диагностические методы
- или XXXV)
- Аномалии числа хромосом[править | править код]
- Признаки
- Выявление хромосомных аномалий
- Ранние признаки
- Также в разделе
- Симптомы[править | править код]
- Клиническая картина синдрома
Генетические и хромосомные заболевания плода
Патологий, возникающих в период внутриутробного развития плода и основывающихся на клеточных мутациях достаточно много. Одни из них встречаются очень редко и диагностируются в сотых, а то и тысячных процентах случаев, другие отличаются постоянством. Мы же рассмотрим те, которые в последние десятки лет все чаще «радуют» своим присутствием в акушерской практике.
К хромосомным и генетическим патологиям плода относят синдромы:
- Дауна — проблемы в 21 хромосоме;
- Патау — патология 13 пары хромосом;
- Эдвардса — нарушения в 18 хромосоме;
- полиплоидия.
В отдельную группу относят трисомии половых хромосом:
- синдром Тернера;
- синдром Клайнфельтера;
- трисомия по Х или Y-хромосоме.
Некоторые заболевания приводят еще к внутриутробной гибели эмбриона, другие несовместимы с нормальным функционированием организма уже родившегося малыша и также заканчиваются смертью на первом году жизни. Ввиду этого, вопрос о целесообразности сохранения на ранних сроках беременности с наличием патологий остается открытым и актуальным, однако, окончательное решение в любом случае остается за семейной парой или женщиной.
Причины мутаций хромосом
Побеспокоиться о здоровье своих детей нужно еще до зачатия, так как патологии хромосом начинаются в момент образования зиготы (слияние сперматозоида с яйцеклеткой). Проконтролировать этот процесс не представляется возможным, так как его специфика плохо изучена.
Для предотвращения наследственных заболеваний врачи настоятельно рекомендуют не пренебрегать этапом подготовки к беременности. Пара должна выявить текущее состояние своего здоровья, вместе с врачом проанализировать анамнез и всех ближайших родственников, оценить свои условия проживания. При плохих результатах врач обязательно расскажет о рисках. В таких случаях паре предлагают искусственное оплодотворение спермой донора (если существует риск передачи болезни по отцовской линии) или суррогатное материнства (при наследственных болезнях по женской линии или от рода матери).
Последние исследования подтверждают связь между генными мутациями и следующими факторами:
- родители старше 35 лет;
- факт патологий в роду;
- неблагоприятные рабочие условия или условия проживания.
Эти факторы повышают риск возникновения хромосомной аномалии. Если пара подтверждает все из них, врачи не рекомендуют зачатие. Когда беременность уже наступила, медицина способна только выяснить степень поражения, определить шансы на выживание и уровень жизни ребенка.
Лечение[править | править код]
Пациенты обычно должны наблюдаться у эндокринолога. При наличии гипогонадизма лечение тестостероном следует рассматривать у всех людей, независимо от когнитивных способностей, из-за положительного влияния на здоровье костей, мышечный тонус, усталость и выносливость, а также с возможными преимуществами для психического здоровья / поведения.
У большинства детей с XXYY наблюдаются некоторые задержки в развитии и трудности в обучении. Следовательно, эти аспекты должны наблюдаться: психология (когнитивное и социально-эмоциональное развитие), логопедия, трудотерапия и физиотерапия. Следует организовать консультации с развивающими педиатрами, психиатрами или неврологами для разработки плана лечения, включающего терапию, поведенческие вмешательства, образовательную поддержку и психотропные препараты для поведенческих и психиатрических симптомов. Общие диагнозы, такие как неспособность к обучению, СДВГ, расстройства аутистического спектра, перепады настроения, тиковые расстройства и другие проблемы психического здоровья, должны быть рассмотрены, обследованы и пролечены. В этой группе наблюдаются хорошие реакции на стандартные медикаментозные методы лечения от невнимательности, импульсивности, тревоги и нестабильности настроения, и такое лечение может положительно влиять на успеваемость, эмоциональное благополучие в долгосрочной перспективе. Плохая координация мелкой моторики может сделать письмо медленным и трудоемким занятием, и трудотерапия и клавиатура должны быть введены в раннем возрасте, чтобы облегчить школьную работу и навыки самопомощи. Образовательные трудности должны оцениваться с помощью полной психологической оценки, чтобы выявить расхождения между устными и служебными навыками и выявить индивидуальные академические потребности. Языковые навыки часто страдают в течение всей жизни, и в зрелом возрасте могут потребоваться вмешательства в области логопедической терапии, направленные на развитие выразительных языковых навыков, диспраксии и др. Адаптивные навыки представляют собой затруднения, требующие поддержки на уровне сообщества почти для всех людей в зрелом возрасте. Могут потребоваться дополнительные рекомендации по лечению, основанные на индивидуальных сильных и слабых сторонах синдрома XXYY.
Профилактика генетических патологий плода
Конечно, превентивные меры в данном случае достаточно условны и все равно не исключают риск аномалий развития плода, но оказать существенное влияние на появление хромосомных патологий женщина может. Условно, попробуем разбить профилактические меры на три типа.
К первоочередным отнесем все мероприятия, направленные на улучшение качества среды обитания, отказ от вредных привычек, обязательное планирование беременности.
Следующий, довольно спорный пункт — это своевременное прерывание беременности, при которой диагностируется патология развития плода.
Наиболее оптимистичными являются профилактические меры третьего типа, когда путем хирургического вмешательства, при условии не серьезных патологий, устраняются все или большая доля дефектов развития.
Такая щепетильная тема, которую мы сегодня затронули, ни в коем случае не должна пугать женщину, формируя у нее стойкое паническое состояние и отказ от беременности после 30-35 лет. Мы лишь призываем к тому, чтобы решение о рождении малыша было осознанным, спланированным и ответственным. Всегда лучше пройти полный комплекс обследований и исключить возможные патологии. В любом случае, сохраняйте положительный настрой и пусть ваши мысли будут только о хорошем, ведь они имеют свойство материализовываться.
Можно ли вылечить или предотвратить хромосомные аномалии

По данным обследования врач вместе с родителями принимает решение о продлении или прерывании текущей беременности. Если патология предполагает возможность вмешательства, может быть проведена реабилитация плода ещё на стадии внутриутробного развития, в том числе и устраняющая порок операция.
Будущие родители ещё на стадии планирования беременности могут посетить генетическую консультацию, которая существует почти в каждом городе. Это особенно необходимо если в роду одного или обоих есть родственники с тяжёлыми наследственными заболеваниями. Генетик составит их родословную и порекомендует исследование кариотипа – полного набора хромосом.
Врачи считают, что такой анализ генов необходим каждой паре, планирующей появление малыша. Это малозатратный универсальный и быстрый метод, позволяющий определить наличие большинства хромосомных болезней любого типа. Будущим родителям всего лишь потребуется сдать кровь. Тем, у кого уже есть в семье ребёнок с генетическим заболеванием, сделать это необходимо в обязательном порядке перед повторной беременностью.
Хромосомные аномалии плода. Признаки
Признаки наличия хромосомной аномалии (ХА) плода во время беременности:
- угроза выкидыша или, как минимум, тянущие боли внизу живота с ранних сроков беременности и на протяжении всей беременности,
- низкий уровень АФП и РАРР-А и повышение ХГЧ на сроке 12-14 недель,
- шейная плода складка более 2 мм на сроке 12 недель,
- малая активность плода (шевеления),
- увеличение лоханок почек по УЗИ на сроках 20-22 недели,
- отставание роста трубчатых костей, начиная с 20-22 недели,
- ранее старение плаценты,
- гипоплазия плаценты,
- гипоксия плода,
- плохие показатели допплерометрии и КТГ,
- маловодие/ многоводие.
Каждый из этих признаков в отдельности и даже все вместе могут быть вариантами нормы.
Виды хромосомных мутаций
Хромосомная мутация – это самопроизвольно произошедшая аномалия с отдельной хромосомой либо с участием нескольких из них. Произошедшие изменения бывают:
- внутри единичной хромосомы, их называют внутрихромосомными;
- межхромосомными, когда отдельные хромосомы обмениваются между собой определёнными фрагментами.
Что может происходить с носительницей информации в первом случае? В результате утраты хромосомного участка происходит нарушение эмбриогенеза и возникают различные аномалии, приводящие к умственному недоразвитию ребёнка или физическим уродствам (пороки сердца, нарушение строения гортани и других органов). Если происходит разрыв хромосомы, после которого вырванный фрагмент встраивается на своё место, но уже перевёрнутым на 180° – говорят об инверсии. Порядок расположения генов меняется. Ещё одна внутрихромосомная мутация – дупликация. В её процессе происходит удвоение участка хромосомы или он дублируется несколько раз, что приводит к множественным порокам умственного и физического развития.
Если же две хромосомы обмениваются фрагментами, явление носит название «реципрокной транслокации». Если фрагмент одной хромосомы встраивается в другую, это называют «нереципрокной транслокацией». «Центрическим слиянием» называют соединение пары хромосом в районе их центромер с утратой соседних участков. При мутации в виде поперечного разрыва соседних хромосом их называют изохромосомами. Такие изменения не имеют внешних проявлений у родившегося потомства, но делает его носителем аномальных хромосом, что может повлиять на возникновение отклонений у следующих поколений. Все типы хромосомной мутации закрепляются в генах и передаются по наследству.
2.Синдром Патау
Частота
встречаемости 1: 6000 новорождённых.
Различают три формы:
—
простая трисомия по хромосоме 13 (75 %
случаев);
—
транслокация (чаще робертсоновская)
(20 % случаев);
—
мозаичная (5%).
Фенотипы:
микроцефалия, тригоноцефалия (расширение
черепа в затылочной и сужение в лобной
части), узкие глазные щели, широкое
основание носа, низко посаженные
деформированные уши, микрофтальмия
(малые размеры глазного яблока),
микрогнатия (малые размеры верхней
челюсти), расщелина губы и нёба,
полидактилия, пороки внутренних органов
(головного мозга, сердца и сосудов,
почек, пищеварения, половых органов).
Дети погибают обычно в течение первых
трёх месяцев жизни.
Анализ на кариотип супругов
Вступая в брак многие пары сталкиваются с проблемой зачатия. Для решения репродуктивных проблем показан цитогенетический анализ. Кариотипирование супругов позволяет выявить аномалии в строении генома, которые мешают завести детей или нарушают процесс вынашивания. Изменить кариотип невозможно, но благодаря диагностике, можно установить истинные причины бесплодия и прерывания беременности, найти пути их решения.
Хромосомный микроматричный анализ проводится для выявления отклонений структуры строения и числа нитей ДНК, которые могут быть причиной наследственных заболеваний у будущего ребенка или бесплодия супругов. Существуют международные стандарты для проведения анализа у будущих родителей:
- Хромосомные патологии в роду, в семье.
- Невынашивание беременности в анамнезе.
- Возраст беременной старше 35 лет.
- Длительные мутагенные воздействия на организм.
На сегодняшний день используют такие методы кариотипирования:
- Анализ хромосом в клетках крови.
Позволяет выявить случаи бесплодия, когда шанс завести ребенка существенно снижен или полностью отсутствует у одного из супругов. Также обследование определяет риск нестабильности генома. Для лечения отклонений пациентам могут быть назначены антиоксиданты и иммуномодуляторы, которые снижают сбои зачатия.
Для исследования проводят забор венозной крови. Из биологической жидкости выделяют лимфоциты, которые стимулируют в пробирке, обрабатывают специальным веществом, окрашивают и изучают. К примеру, при синдроме Клайнфельтера, который проявляется мужским бесплодием, в кариотипе присутствует лишняя хромосома 47 ХХ. Также могут быть выявлены структурные изменения генома: инверсия, делеция, транслокация.
- Пренатальное исследование.
Определяет хромосомные патологии плода на ранних сроках беременности. Такое исследование необходимо для диагностики генетических заболеваний или пороков развития, которые приводят к внутриутробной гибели плода.
Для проведения исследования могут быть использованы такие методы:
- Неинвазивные – безопасны для матери и плода. Диагностику проводят с помощью УЗИ ребенка и развернутого биохимического анализа крови женщины.
- Инвазивные – биопсия хориона, кордоцентез, плацентоцентез, амниоцентез. Для анализа проводят забор клеток плаценты или хориона, околоплодных вод или крови из пуповины. Несмотря на высокую точность диагностики, инвазивные методики имеют повышенный риск осложнений, поэтому проводятся только по строгим врачебным показаниям: выявленные во время УЗИ патологии плода, роженица старше 35 лет, родители с хромосомными аномалиями, изменение биохимических маркеров крови.
Для цитогенетического исследования может быть использована не только кровь, но и эякулят. Данный метод называется Tunel и позволяет определить одну из самых распространенных причин мужского бесплодия при условии нормального кариотипа – фрагментацию ДНК сперматозоидов.
При обнаружении генных мутаций или хромосомных аберраций у одного из супругов, врач рассказывает о возможных рисках и вероятности рождения ребенка с отклонениями. Поскольку генные патологии неизлечимы, то дальнейшее решение супруги принимают самостоятельно: воспользоваться донорским материалом (сперма, яйцеклетка), рискнуть родить или остаться без детей.
Если отклонения в геноме выявлены в процессы вынашивания, причем как у женщины, так и у эмбриона, то врачи рекомендуют прерывать такие беременности. Это связано с повышенным риском рождения малыша с серьезными, а в некоторых случаях и несовместимыми с жизнью отклонениями. Проведением анализов и расшифровкой их результатов занимается врач-генетик.
[], [], [], [], []
Синдром кошачьего крика — хромосомная аномалия у детей
Синдром кошачьего крика возникает в результате аномалии пятой хромосомы.
Основные признаки хромосомной аномалии
Синдром проявляется пронзительным криком новорожденного, напоминающего кошачий. Он обусловлен изменением гортани: сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки. Обычно наблюдается глубокая умственная отсталость.
Многие больные с хромосомной аномалией доживают до взрослого состояния. Продолжительность жизни зависит от тяжести пороков внутренних органов. С возрастом почти полностью исчезает «кошачий крик«, мышечная гипотония, лунообразное лицо, но прогрессирует умственная отсталость, косоглазие.
Риск передачи по наследству этого заболевания составляет менее 1 %.
Теперь вы знаете основные причины хромосомной аномалии у детей, а также признаки хромосомной аномалии у ребенка. Здоровья вашим детям!
Диагностические методы
Самый информативный метод диагностики хромосомных патологий плода — первый скрининг (его ещё называют двойным тестом). Делают в 12 недель беременности. Он включает в себя:
- УЗИ (выявляются маркеры, обозначенные выше);
- анализ крови (берётся из вены на голодный желудок), показывающий уровень АФП, ХГЧ, АРР-А.
Следует понимать, что данный анализ на хромосомные патологии плода не может дать точного, 100% подтверждения или опровержения наличия аномалий. Задача врача на данном этапе — рассчитать риски, которые зависят от результатов исследований, возраста и анамнеза молодой мамы. Второй скрининг (тройной тест) ещё менее информативен. Самая точная диагностика — это инвазивные методы:
- биопсия хориона;
- забор пуповинной крови;
- анализ амниотической жидкости.
Цель всех этих исследований — определить кариотип (совокупность признаков набора хромосом) и в связи с этим хромосомную патологию. В этом случае точность постановки диагноза составляет до 98%, тогда как риск выкидыша — не более 2%. Как же происходит расшифровка данных, полученных в ходе этих диагностических методик?
или XXXV)
Проявляется у лиц мужского пола в период полового созревания и характеризуется атрофией яичек, гиалинизацией семенных канальцев, бесплодием и, часто, гинекомастией. Частота синдрома Кляйнфельтера составляет 1: 500 мужчин. Клетки содержат 47 хромосом с дополнительной Х-хромосомой (генотип ХХУ) вследствие нерасхождения ХХ гомологичных хромосом. Тельца полового хроматина оказываются в 80% случаев. Иногда больные с синдромом Кляйнфельтера имеют 48 хромосом, то есть 44 аутосомы и 4 половые хромосомы (ХХХУ). Синдром обычно не сопровождается задержкой умственного развития, но чем больше Х хромосом у генотипе, тем больше риск развития умственной отсталости.
Аномалии числа хромосом[править | править код]
Болезни, обусловленные нарушением числа хромосом в клетках человека
- синдром Дауна — трисомия по 21-й хромосоме (или наличие дополнительных копий генетического материала этой хромосомы по другим причинам — за счёт транслокации или дупликации);
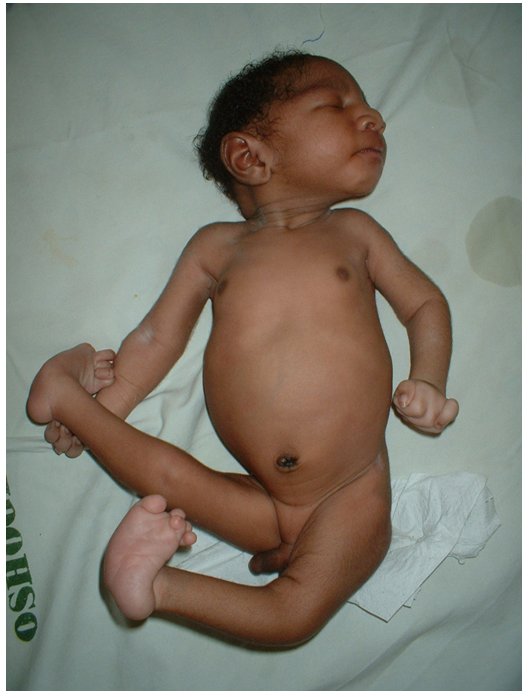
- синдром Патау — трисомия по 13-й хромосоме, характеризуется множественными пороками развития, идиотией, часто — полидактилия, нарушения строения половых органов, глухота; большинство больных не доживают до одного года;
- синдром Эдвардса — трисомия по 18-й хромосоме, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы; 60 % детей умирают в возрасте до 3 месяцев, до года доживают лишь 10 %, основной причиной служит остановка дыхания и нарушение работы сердца.
Болезни, связанные с нарушением числа половых хромосом
- Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 Х0) вследствие нарушения расхождения половых хромосом; к признакам относится низкорослость, половой инфантилизм и бесплодие, различные соматические нарушения (микрогнатия, короткая шея и др.);
- полисомия по Х-хромосоме — включает трисомию (кариотип 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижение интеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения;
- полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотип 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы;
- Синдром Клайнфельтера — полисомия по X-хромосомам у мальчиков (47, XXY), признаки: евнухоидный тип сложения, гинекомастия, слабый рост волос на лице, в подмышечных впадинах и на лобке, половой инфантилизм, бесплодие; умственное развитие отстает, однако иногда интеллект нормальный.
Болезни, причиной которых является полиплоидия
триплоидии, тетраплоидии и т. д.; причина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69, XXX); почти всегда летальны до рождения.
Признаки
Так как процесс возникновения и развития подобного рода отклонений изучен недостаточно, маркеры хромосомной патологии плода считаются условными. К ним относятся:
- угроза выкидыша, тянущие боли в нижней части живота на ранних сроках беременности;
- низкий уровень РАРР-А (протеин А из плазмы) и АФП (белок, вырабатываемый организмом эмбриона), повышенный ХГЧ (хорионический гонадотропин — гормон плаценты): для получения таких данных берётся из вены кровь на хромосомную патологию плода на сроке 12 недель (+/- 1-2 недели);
- длина носовых костей;
- увеличенная шейная складка;
- неактивность плода;
- увеличенные лоханки почек;
- замедленный рост трубчатых костей;
- ранее старение или гипоплазия плаценты;
- гипоксия плода;
- плохие результаты допплерометрии (метода УЗИ для выявления патологий кровообращения) и КТГ (кардиотокографии);
- мало— и многоводие;
- гиперэхогенный кишечник;
- маленький размер верхнечелюстной кости;
- увеличенный мочевой пузырь;
- кисты в головном мозге;
- отёчности в области спины и шеи;
- гидронефроз;
- лицевые деформации;
- кисты пуповины.
Неоднозначность этих признаков в том, что каждый из них в отдельности, как и весь выше перечисленный комплекс, может быть нормой, обусловленной индивидуальными особенностями организма матери или ребёнка. Самые точные и достоверные данные дают обычно анализ крови на хромосомные патологии, УЗИ и инвазивные методики.
Выявление хромосомных аномалий
Чаще всего будущих родителей интересует вопрос, можно ли диагностировать хромосомные патологии у плода в период беременности? Благодаря современным технологиям, диагностировать хромосомные аномалии во время беременности вполне реально. Существуют определенные маркеры, по которым можно предположить наличие патологии у плода еще в период вынашивания. Итак, маркеры хромосомных аномалий:
- Тянущие боли внизу живота, особенно на самых ранних сроках беременности.
- Угроза выкидыша, как на ранних, так и на поздних сроках.
- Высокий уровень ХГЧ (хорионический гонадотропин человека) на 12-14 недели беременности.
- По данным УЗИ — шейная складка у плода более 2 мм на сроке 12 недель, большие размеры лоханок почек (20-22 недели), замедленный рост трубчатых костей.
- Нет ощущений шевеления плода.
- Патологии плаценты — раннее старение, гипоплазия плаценты, гипоксия плода.
- Патологические показатели по данным допплерографии или кардиотокографии.
- Большое или наоборот недостаточное количество околоплодных вод.
Но стоит помнить о том, что данные маркеры не являются стопроцентными признаками хромосомной патологии, и могут быть вариантом нормы.
Этапы диагностики
Диагностика хромосомных аномалий состоит из нескольких этапов, два из которых являются обязательными скрининговыми исследованиями для всех беременных. А третий этап является инвазивным, и показан только при наличии высокого риска по данным неинвазивных методик, поскольку сопряжен с рядом осложнений.

Итак, первый этап — это двойное исследование, которое включает УЗИ и лабораторные исследования АФП, РАРР-А и ХГЧ крови. Первый скрининг нужно проводить на 12 неделе беременности. Второй этап — это те же исследования, но уже во втором триместре.
Данные методики не дают ответ на вопрос, есть у плода хромосомная аномалия или нет, а лишь определяет степень риска. Наиболее точные сведения возможно получить только при применении инвазивных методик исследования, которые включают биопсию хориона, забор пуповинной крови, исследование амниотической жидкости. Они позволяют определить кариотип плода, и дают ответ с вероятностью до 98%.
Нужно учитывать тот факт, что инвазивные методики увеличивают риск выкидыша. Выбор проводить инвазивные исследования или нет, будет зависеть от многих аспектов. Принимать решение следует, взвесив все риски и возможные исходы, а также, какое именно заболевание предполагают у плода, какой прогноз для ребенка в случае рождения с такой патологией, готовы ли будущие родители к рождению ребенка с хромосомной аномалией.
Ранние признаки
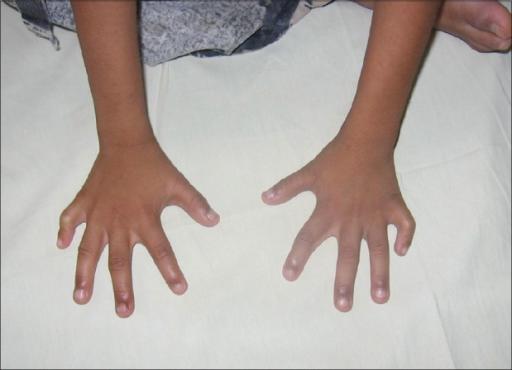
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается. Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания. В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит. В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Также в разделе
| Значение генетики для медицины Прогресс в развитии медицины и общества приводит к относительному возрастанию доли генетически обусловленной патологии в заболеваемости, смертности,… | |
| Синдром частичной трисомии по короткому плечу хромосомы 9 (9р+) Синдром частичной трисомии по короткому плечу хромосомы 9 (9р+) — наиболее частая форма частичных трисомии (опубликовано около 200 сообщений о больных с такой… | |
| Миодистрофия Дюшенна-Беккера Это одна из частых форм многочисленных наследственных нервно-мышечных заболеваний. Мышечные дистрофии характеризуются прогрессирующими дегенеративными… | |
| Наследственность Любые проявления жизнедеятельности организма являются результатом взаимодействия наследственных и средовых факторов. Болезнь также развивается на основе… | |
| Синдром Коккейна Синдром Коккейна — редкое генетическое заболевание, описанное Коккейном в 1936 г. Проявляется умственной отсталостью, атрофией зрительного нерва, карликовостью,… | |
| Клиническая диагностика наследственных болезней Большинство наследственных болезней имеет хроническое течение, вследствие чего повторная обращаемость при таких болезнях высокая. Особенно много больных с… | |
| Митохондриальная наследственность Митохондрии передаются с цитоплазмой яйцеклеток. Спермии не имеют митохондрий, поскольку цитоплазма элиминируется при созревании мужских половых клеток. В… | |
| Наследственная предрасположенность к алкоголизму Алкоголизм обычно формируется при наличии определённых факторов генетической и биохимической предрасположенности. Наследственная предрасположенность к… | |
| Адреногенитальный синдром Синоним: врождённая вирилизующая гиперплазия коры надпочечников. Адреногенитальный синдром относится к группе наследственных нарушений биосинтеза… | |
| Синдром Блума Синдром Блума (врожденная телеангиэктатическая эритема) — наследственный симптомокомплекс, характеризующийся телеангиэктатической эритемой лица, малым… |
Симптомы[править | править код]
Симптомы 49, XXXXY схожи с симптомами синдрома Клайнфелтера и 48, XXXY, однако они обычно намного более выражены при синдроме 49, XXXXY. Анеуплоидия часто приводит к летальному исходу, но в этом случае существует «инактивация Х», при которой эффект дополнительных Х хромосом значительно снижается.
Репродуктивное
Лица с синдромом 49, XXXXY, как правило, демонстрируют неразвитые вторичные половые признаки и стерильность.
Гипопластические гениталии.
Физические
Мужчины с таким кариотипом как правило, имеют многочисленные скелетные аномалии. Эти скелетные аномалии включают в себя:
- Genu Valgum
- Pes Cavus
Клинодактилия
Также:
Психические
Как и при синдроме Дауна, психические проявления синдрома 49, XXXXY различны. Типичными являются нарушения речи и неадекватные поведенческие проблемы. В одном исследовании рассматривались мужчины с диагнозом 48, XXYY, 48, XXXY и 49, XXXXY. Они обнаружили, что мужчины с 48, XXXY и 49, XXXXY функционируют на гораздо более низком когнитивном уровне, чем мужчины их возраста. Эти мужчины также имеют тенденцию проявлять более незрелое поведение для своего возраста; повышенные агрессивные тенденции были также процитированы в этом исследовании.
Патофизиология
Как видно из названия, у человека с этим синдромом есть одна Y-хромосома и четыре X-хромосомы на 23-й паре, таким образом, имеется сорок девять хромосом, а не нормальные сорок шесть. Как и в большинстве категорий анеуплоидных расстройств, синдром 49, XXXXY часто сопровождается умственной отсталостью. Его можно рассматривать как форму или вариант синдрома Клайнфелтера (47, XXY). Люди с этим синдромом, как правило, мозаичные, 49, XXXXY / 48, XXXY.
Это генетическое, но не наследственное заболевание, что означает, что, хотя гены родителей вызывают синдром, существует небольшая вероятность того, что синдром у более чем одного ребенка. Вероятность наследования заболевания составляет около одного процента.
Клиническая картина синдрома
Обычно дети с синдромом Шерешевского-Тернера, рождаются раньше срока. Но даже в случае беременности, доношенной до 40 недель, вес новорожденного редко достигает 3 кг, а рост более 50 см. Сразу же после родов, можно заметить у ребенка признаки, которые характерны для заболевания:
- Укороченная шея;
- Птеригиум-синдром – кожные складки в виде крыльев на боках шеи;
- Нарушение оттока лимфы;
- Отеки на стопах и кистях;
- Наличие врожденных пороков сердечно-сосудистой системы.
Далее на первый план выступают проблемы с кормлением – у детей нарушено сосание, они часто срыгивают «фонтаном», находится в моторном возбуждении. На первых годах жизни можно заметить отставание в развитие – ребенок поздно начинает сидеть, ходить, говорить. Также для синдрома Шерешевского-Тернера характерно частое, повторное возникновение среднего отита, которое впоследствии приводит к кондуктивной тугоухости.
На момент полового созревания люди с синдромом Шерешевского-Тернера выглядят следующим образом: рост людей редко когда превышает 150 см. Кроме этого, они имеют характерный внешний вид:
- Лицо обретает определенное выражение – «лицо Сфинкса»;
- Шея укорочена, присутствует птеригиум-синдром;
- Граница роста волос занижена;
- Челюсть недоразвита – микрогнатия;
- Уши увеличены, часто бывает лопоухость;
- Грудная клетка слишком широкая.
Жизнь с синдромом Шерешевского-Тернера непроста, при данной патологии значительно поражается костная система организма. Часто возникает сколиоз, дисплазия суставов, особенно тазобедренных, девиация локтей. На черепе наблюдается микрогнатия, неправильный прикус и готическое небо. В связи с недостаточным количеством эстрогена, люди с таким синдромом подвержены раннему возникновению остеопороза. У них часто случаются переломы позвоночника, костей кисти и шейки бедра.
Сколько живут люди с синдром Шерешевского-Тернера зависит от степени тяжести заболевания и от осложнений систем органов, которые возникают вследствие данной патологии . Со стороны сердечно-сосудистой системы встречаются такие пороки, как коарктация и аневризма аорты, дефекты межжелудочковой и межпредсердной перегородки. В почках часто бывает раздвоение лоханок, стеноз артерий, подковообразная форма самой почки. Подобные патологии приводят к артериальной гипертензии. У больных синдромом Шерешевского-Тернера нередко развивается косоглазие, близорукость и опущение века. К тому же они могут также страдать и дальтонизмом.
Умственные способности, как правило, сохраняются на должном уровне, но в некоторых случаях может наблюдаться олигофрения. Людей, страдающих синдромом Шерешевского-Тернера, часто сопровождают соматические заболевания – алопеция, микседема, витилиго, дефицит ферментов в тонком кишечнике, ожирение. Практически во всех случаях диагностируется диабет первого или второго типа и ишемическое поражение сердца. Доказано, что у таких пациентов рак толстого кишечника возникает в несколько раз чаще, чем у здоровых людей.
Практически у 100% больных девушек диагностируется первичный гипогонадизм. Внутренние половые органы недоразвиты – матка гипоплазирована, а вместо яичников с двух сторон находятся фиброзные тяжи. Женщины с синдромом Шерешевского-Тернера обычно полностью стерильны – в яичниках отсутствуют фолликулы с яйцеклетками. Вульва видоизменена: большие губы похожи на мошонку, а малые и клитор недоразвиты. Девственная плева может отсутствовать. Некоторые задаются вопросом как приходят месячные с синдромом Шерешевского-Тернера – в период полового созревания обнаруживается первичная аменорея. Грудные железы развиты недостаточно, наблюдается незначительное оволосение в участке лобка и подмышек. Беременность, которая возникла природным путем, возможно только в случае мозаичной формы поражения X-хромосомы.
Половые органы мужчины также недоразвиты. Яички гипоплазированы, часто не опускаются в мошонку, иногда диагностируется анорхия – полное отсутствие ткани яичек в организме. Наблюдается сильно заниженный уровень мужского гормона – тестостерона.