Синдром эдвардса
Содержание:
- Лечение синдрома Эдвардса
- Причины заболевания
- Проявления синдрома
- Прогноз
- Частота появления
- Вариации
- Болезнь Эдвардса на УЗИ
- Синдром Эдвардса – прогноз
- Проявления синдрома
- Влияние трисомии 18 на ребенка
- Симптомы
- Диагностика
- Клиническая картина синдрома
- Диагностика
- Лечение
- Первые признаки синдрома Эдвардса
- Признаки синдрома Эдвардса
Лечение синдрома Эдвардса
Нужно учитывать, что только один из десяти детей, родившихся с подобной патологией, доживают до одного года. Потому первоначальное лечение состоит в избавлении от заболеваний, повышающих риск летального исхода. Если имеет место быть атрезия кишечника или анального прохода, используются меры для дефекации. Ребенка кормят через зонд. Если в организме новорождённого наблюдается активные воспалительные или инфекционные процессы, осуществляется терапевтическое лечение.
Если ребёнка всё же удаётся спасти, в последующем может быть выполнено оперативное вмешательство. В первую очередь выполняется устранение волчьей пасти. Дополнительно проходит борьба с пороками сердца. Пупочная и паховая грыжа удаляются при помощи оперативных методов.
Проводится и медикаментозная терапия. Ребенку назначают лекарства, которые позволяют избавить его от запоров, повышенной кислотности и метеоризма. Родители должны понимать, что синдром Эдвардса способствует появлению следующих патологий:
- отит;
- новообразование в почках;
- гипертензия легких;
- конъюнктивит;
- воспаление лёгких;
- высокое артериальное давление;
- синусит и фронтит;
- инфекционные заболевания мочеполовой системы.
Из-за большого количества патологических изменений, происходящих во внутренних и внешних органах больного, дальнейший прогноз развития неблагоприятен. Те дети, которые сумеют дожить до года и достичь относительно взрослого возраста, будут иметь явное умственное торможение в развитии. Они не смогут самостоятельно ухаживать за собой и удовлетворять свои естественные требования. За такими пациентами необходимо обеспечить постоянный уход и контроль на протяжении всей их жизни.
Однако дети с отклонениями понимают, когда с ними ласково общаются, играют и утешают. Дети с таким синдромом могут научиться самостоятельно есть, улыбаться. Возможно постепенное обучение другим полезным бытовым навыкам.
Высокое количество патологий внутренних органов, развившихся неправильно, приводит к высокому проценту детской смертности. Если в первые месяцы беременности женщина попала в группу риска по этому заболеванию, медики рекомендует выполнить аборт по медицинским показаниям. Однако окончательное решение остается за женщиной.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
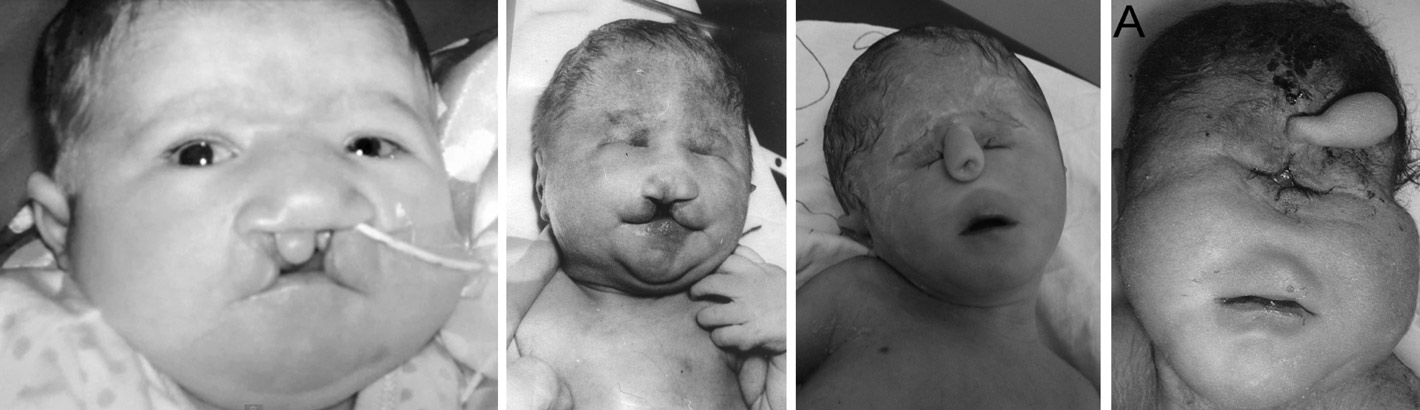
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
Болезнь Эдвардса на УЗИ
Всем беременным женщинам предписывается пройти обследование на предмет наличия хромосомных отклонений у плода. Синдром Эдвардса до 12 недель никак не диагностируется, хотя отклонения есть с момент оплодотворения яйцеклетки, но начиная с 12 недели возможно обнаружить характерные для этого заболевания симптомы. К ним относятся:
- Брадикардия (нарушение у плода сердцебиения в виде снижения частоты сердечных сокращений),
- Омфалоцеле (наличие в брюшной полости грыж),
- Отсутствие визуально определенных косточек носа,
- Отсутствие в пуповине одной артерии (в норме в пуповине должно быть две артерии),
- Наличие кисты сосудистых сплетений (киста сама по себе опасности для плода не несет и самопроизвольно исчезает к 26 неделе беременности, но она часто возникает на фоне различных генетических заболеваний у плода, в том числе синдрома Эдвардса).
Наличие хотя бы одного из указанных отклонений выступает поводом для направления беременной женщины на дополнительное исследование. Принудительного исследования женщины не проводится, на все диагностические процедуры и манипуляции она должна давать добровольное согласие
Врач обязан разъяснить смысл и важность диагностических процедур в данном случае, а также порядок их проведения
На более поздних сроках беременности на УЗИ могут быть обнаружены расщепление позвоночника плода вследствие дефекта нервной трубки, увеличение жидкости в воротниковом пространстве, укорочение костей, деформация черепа, изменения в структурах мозга. Эти отклонения свойственным многим хромосомным аномалиям.
УЗИ на выявление хромосомных заболеваний у плода называется скрининговым и проводится врачами, прошедшими специальную подготовку в области генетических аномалий развития. Для этого женщина обычно направляется в иное учреждение для обследования, поскольку не в каждой поликлинике есть необходимое оборудование и специалист с соответствующим уровнем подготовки.
ДополнительноПрограмма скрининга беременных в 1 триместре включает в себя также анализ сыворотки крови, поскольку ни один самый точный прибор УЗИ не может с абсолютной точностью показать наличие или отсутствие заболеваний у плода.
Риск рождения ребенка с хромосомным заболеванием рассчитывается по таким показателям:
- количество ассоциированного с беременностью плазменного протеина А (анализ сыворотки)
- количество свободной β-субъединицы хорионического гонадотропина человека (анализ сыворотки),
- морфологические признаки (УЗИ).
Индивидуальный риск рождения ребенка с генетической аномалией развития 1/100 и выше.
Самый точный диагноз может быть поставлен на основе исследования плодного материала.
Для этого проводится инвазивная процедура. Какая именно процедура будет проведена, зависит от срока беременности:
- В 8-12 недель биопсия ворсин хориона (ткань хориона имеет тот же набор хромосом, что и плод, потому с точностью показывает наличие либо отсутствие генетических отклонений,
- В 14-18 недель амниоцентез (забор и анализ околоплодной жидкости),
- В 18-20 недель и позднее кордоцентоз (забор и анализ пуповинной крови).
Амниоцентез и кордоцентез часто проводятся вместе в ходе одной диагностической процедуры. Это позволяет получить более всесторонние и точные результаты.
Важно В случае постановки диагноза хромосомных нарушений в том числе и синдрома Эдвардса у плода и врожденных пороков развития с неблагоприятным прогнозом для жизни и здоровья ребенка по решению перинатального консилиума и добровольного согласия беременной женщины производится прерывание беременности независимо от срока беременности. Женщина обязательно должна быть проинформирована о диагнозе плода, о течении диагностированного заболевания, особенностях развития и качестве жизни детей, рожденных с подобной аномалией
Женщина обязательно должна быть проинформирована о диагнозе плода, о течении диагностированного заболевания, особенностях развития и качестве жизни детей, рожденных с подобной аномалией.
После 16-18 недель беременности проводится тройной скрининговый тест на хромосомные отклонения плода. В ходе биохимического анализа крови оцениваются такие показатели:
- α-фетопротеин (альфа-ФП);
- свободная β-субъединица хорионического гормона человека (бета-ХГЧ);
- эстриол свободный.
Синдром Эдвардса – прогноз
Большинство зародышей с описываемой генетической аномалией гибнет еще во время вынашивания из-за отторжения организмом неполноценного плода. После рождения прогноз тоже неутешителен. Если диагностирован синдром Эдвардса, сколько живут такие дети, рассмотрим в процентных соотношениях:
- 60% – не более 3-х месяцев;
- 7-10% – 1 год;
- около 1% – до 10 лет.
В исключительных случаях (частичная или мозаичная трисомия 18) единицы могут достичь зрелости. Даже в таких ситуациях синдром Джона Эдвардса будет неумолимо прогрессировать. Повзрослевшие дети с данной патологией навсегда остаются олигофренами. Максимум, чему их можно научить:
- поднимать голову;
- улыбаться;
- самостоятельно есть;
- узнавать ограниченный круг людей.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Влияние трисомии 18 на ребенка
Дополнительный генетический материал вызвает множество проблем (врожденные дефекты) у растущего ребенка в утробе матери и после появления на свет. Так же, как с синдромом Дауна проблемы варьируются от легкой до тяжелой степени.
Каждый ребенок имеет собственный уникальный профиль того, как Trisomy 18 влияет на их развивающееся тело и органы. Общие проблемы:
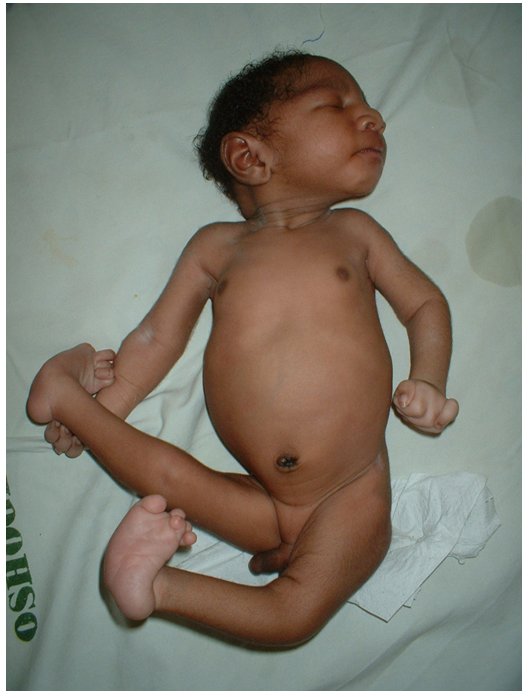
- Сердечные пороки:
- VSD (Желудочковый дефект сепастьяна): отверстие между нижними камерами;
- ASD (дефект седалищного предсердия): отверстие между верхними камерами;
- Коарктация аорты: сужение выходного сосуда;
- Проблемы с почками;
- Часть кишечного тракта находится вне желудка (омфалоцеле);
- Пищевод не соединяется с желудком (пищеводная артезия);
- Избыточная амниотическая жидкость (полигидрамниоз);
- Сжатые руки;
- Карман жидкости на мозге (кисты сосудистого сплетения);
- Ножки качалки;
- Отложенный рост;
- Маленькая челюсть (mycrognathia);
- Маленькая головка (микроцефалия);
- Низко посаженные, несформированные уши;
- Клубничная голова;
- Серьезные задержки развития;
- Пупочная или паховая грыжа.
Симптомы
Семейной паре нужно понимать, что такое болезнь эдвардса, и какие страдания она способна принести родителям и ребенку. Симптоматика крайне не радостна. Первые признаки будут давать о себе знать уже после рождения:
- Асфиксия.
- Волчья пасть.
- Опущение век (птоз).
- Слабоумие.
- Нарушение глотания.
- Нарушение дыхания.
- Сердечные и сосудистые пороки.
- Заболевания системы пищеварения.
- Нарушения работы мочеполовой системы.
- Проблемы с психикой.
Учитывая все перечисленные симптомы, можно подвести итог, что больной ребенок никогда не станет полноценным членом общества, как бы этого не хотелось. Жизненный путь у него будет коротким, наполненным постоянными преградами. При этом больной не будет понимать и осознавать своего тяжелого состояния из-за тяжелой степени олигофрении.
Но это далеко не все проявления. Эдвардс описал свыше ста тридцати тяжелых симптомов данного недуга.
Диагностика
О генетических патологиях чаще всего можно узнать, пока женщина еще вынашивает ребенка. Это касается и трисомий. Скрининг беременности проводят с 11-й по 13-ю неделю. Женщина сдает анализы крови (биохимию), проводится УЗИ. Также диагностика заключается в определении каротипа эмбриона, если женщина находится в группе риска (отягощенный семейный анамнез, инфекционные болезни в первом триместре и т. д.).
В скрининге первого семестра определяют, сколько в крови хорионического гормона человека и плазменного протеина А ассоциированного с беременностью. Потом учитывают возраст беременной, чтобы узнать, с каким риском у нее может родиться ребенок с трисомией 18.
Если женщину отнесли в группу риска, чуть позже делают биопсию плода, чтобы точно знать, родится ли ребенок с отклонениями, или здоровый. С 8 до 12 неделю берется анализ ворсин хориона. С 14 по 18-ю неделю проводится изучение вод, окружающих плод. После 20-й недели могут сделать кордоцентез. Процедура подразумевает, что возьмут крови из пуповины (в процессе применяется ультразвук для контроля взятия материала).
В материале обнаруживают количество хромосом. В этом помогает метод КФ–ПЦР. При условии непрохождения беременной генетического скрининга на поздник сроках гестации делают предварительную диагностику генетической мутации методом ультразвукового исследования. Во втором и третьем триместре есть признаки, которые говорят о том, что ребенок с большой вероятностью родится с трисомией:
- заячья губа
- низко расположенные уши плода
- микроцефалия
- волчья пасть
- пороки опорно–двигательного аппарата
- пороки развития мочеполовой системы
- пороки сердца и сосудов
Клиническая картина синдрома
Обычно дети с синдромом Шерешевского-Тернера, рождаются раньше срока. Но даже в случае беременности, доношенной до 40 недель, вес новорожденного редко достигает 3 кг, а рост более 50 см. Сразу же после родов, можно заметить у ребенка признаки, которые характерны для заболевания:
- Укороченная шея;
- Птеригиум-синдром – кожные складки в виде крыльев на боках шеи;
- Нарушение оттока лимфы;
- Отеки на стопах и кистях;
- Наличие врожденных пороков сердечно-сосудистой системы.
Далее на первый план выступают проблемы с кормлением – у детей нарушено сосание, они часто срыгивают «фонтаном», находится в моторном возбуждении. На первых годах жизни можно заметить отставание в развитие – ребенок поздно начинает сидеть, ходить, говорить. Также для синдрома Шерешевского-Тернера характерно частое, повторное возникновение среднего отита, которое впоследствии приводит к кондуктивной тугоухости.
На момент полового созревания люди с синдромом Шерешевского-Тернера выглядят следующим образом: рост людей редко когда превышает 150 см. Кроме этого, они имеют характерный внешний вид:
- Лицо обретает определенное выражение – «лицо Сфинкса»;
- Шея укорочена, присутствует птеригиум-синдром;
- Граница роста волос занижена;
- Челюсть недоразвита – микрогнатия;
- Уши увеличены, часто бывает лопоухость;
- Грудная клетка слишком широкая.
Жизнь с синдромом Шерешевского-Тернера непроста, при данной патологии значительно поражается костная система организма. Часто возникает сколиоз, дисплазия суставов, особенно тазобедренных, девиация локтей. На черепе наблюдается микрогнатия, неправильный прикус и готическое небо. В связи с недостаточным количеством эстрогена, люди с таким синдромом подвержены раннему возникновению остеопороза. У них часто случаются переломы позвоночника, костей кисти и шейки бедра.
Сколько живут люди с синдром Шерешевского-Тернера зависит от степени тяжести заболевания и от осложнений систем органов, которые возникают вследствие данной патологии . Со стороны сердечно-сосудистой системы встречаются такие пороки, как коарктация и аневризма аорты, дефекты межжелудочковой и межпредсердной перегородки. В почках часто бывает раздвоение лоханок, стеноз артерий, подковообразная форма самой почки. Подобные патологии приводят к артериальной гипертензии. У больных синдромом Шерешевского-Тернера нередко развивается косоглазие, близорукость и опущение века. К тому же они могут также страдать и дальтонизмом.
Умственные способности, как правило, сохраняются на должном уровне, но в некоторых случаях может наблюдаться олигофрения. Людей, страдающих синдромом Шерешевского-Тернера, часто сопровождают соматические заболевания – алопеция, микседема, витилиго, дефицит ферментов в тонком кишечнике, ожирение. Практически во всех случаях диагностируется диабет первого или второго типа и ишемическое поражение сердца. Доказано, что у таких пациентов рак толстого кишечника возникает в несколько раз чаще, чем у здоровых людей.
Практически у 100% больных девушек диагностируется первичный гипогонадизм. Внутренние половые органы недоразвиты – матка гипоплазирована, а вместо яичников с двух сторон находятся фиброзные тяжи. Женщины с синдромом Шерешевского-Тернера обычно полностью стерильны – в яичниках отсутствуют фолликулы с яйцеклетками. Вульва видоизменена: большие губы похожи на мошонку, а малые и клитор недоразвиты. Девственная плева может отсутствовать. Некоторые задаются вопросом как приходят месячные с синдромом Шерешевского-Тернера – в период полового созревания обнаруживается первичная аменорея. Грудные железы развиты недостаточно, наблюдается незначительное оволосение в участке лобка и подмышек. Беременность, которая возникла природным путем, возможно только в случае мозаичной формы поражения X-хромосомы.
Половые органы мужчины также недоразвиты. Яички гипоплазированы, часто не опускаются в мошонку, иногда диагностируется анорхия – полное отсутствие ткани яичек в организме. Наблюдается сильно заниженный уровень мужского гормона – тестостерона.
Диагностика
Поскольку синдром Эдвардса характеризуется довольно большим количеством ярко выраженных отклонений, его довольно просто диагностировать даже по внешним проявлениям. Однако этого недостаточно, чтобы поставить окончательный диагноз.
Диагностика синдрома Эдвардса складывается из трех этапов — обследование супружеских пар до момента зачатия, беременной женщины до родов и ребенка после появления на свет.
Диагностика до зачатия ребенка — идеальный вариант, но не всегда применимый. Специалисты-генетики могут лишь предположить, каков риск рождения ребенка с хромосомным заболеванием в данной семье.
До момента зачатия врачи собирают семейный анамнез, опрашивая родителей об их родословной.
Большое внимание специалисты уделяют факторам риска: возрасту матери, перенесенным инфекционным заболеваниям, хроническим болезням, вредным привычкам.
Генетический анализ родителей – полноценное исследование, с помощью которого составляется их кариотип и обнаруживаются участки ДНК с дефектными генами.
Диагностика в период внутриутробного развития дает более точные результаты, поскольку обследуют организм плода. Пренатальная диагностика — важный этап в процессе выявления хромосомных нарушений.
- Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока – неинвазивные методы, полностью безопасные и рекомендованные всем беременным. Признаки синдрома Эдвардса: отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития черепа и костей, агенезия пупочной артерии, малая величина плаценты, многоводие, брадикардия, отсутствие носовых костей, 2 артерии в пуповине, кисты сосудистых сплетений. Диагностика с помощью ультразвукового исследования является достоверной на 100%.
- Стандартный пренатальный скрининг включает анализ крови на сывороточные маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. В таких случаях показано искусственное прерывание беременности по медицинским показаниям.
- Амниоцентез – клеточный анализ околоплодных вод. Инвазивная методика, осуществляемая путем забора амниотической жидкости шприцем. Ее клетки содержат образцы ДНК плода, которые проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пупочной крови плода, позволяющее определить генетические аномалии с высокой точностью.
- Биопсия хориона представляет собой пункцию матки через переднюю брюшную стенку и забор ткани для анализа – стандартного генетического исследования.
Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики с последующим кариотипированием плода. Инвазивные методы считаются самыми точными и надежными, но требующими оперативного вмешательства и проникновения в оболочку плода. Диагноз подтверждается при помощи определения кариотипа малыша путем КФ-ПЦР.
Диагностика синдрома Эдвардса после рождения самая легкая, быстрая и точная. После выявления некоторых врожденных дефектов проводят генетический анализ для подтверждения диагноза. Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, являются эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение
Лечение синдрома Эдвардса требует комплексного подхода. За ребенком, больным синдромом Эдвардса, необходим постоянный и тщательный уход, необходимо постоянно контролировать поведение ребенка и соблюдать рекомендации врача по уходу за больным. Иногда при явно выраженных пороках необходимо проводить хирургическую коррекцию. Также необходимо лечить симптомы заболевания, для этого врач назначает медикаментозное лечение, которое необходимо тщательно выполнять и соблюдать.
Осложнения
В основном, дети с синдромом Эдвардса погибают в первый год жизни. Наступает летальный исход вследствие нарушения сердечной деятельности или остановки дыхания.
Профилактика
Нет специальных методов профилактики синдрома Эдвардса. Если имеются наследственные заболевания в предыдущих поколениях, то необходимо перед планированием беременности проконсультироваться у врача.
Первые признаки синдрома Эдвардса
Первые признаки развития патологии заметно еще в процессе внутриутробного развития. Во время беременности наблюдается маленькая плацента, а также многоводие. Подвижность плода снижена. Присутствует только единственная пупочная артерия. Ребенок появляется на свет с массой тела чуть больше 2 кг. Дополнительно может присутствовать пренатальная гипотрофия. В некоторых случаях выявляются и другие признаки синдрома Эдвардса. Так, может быть выявлена с фиксацией при рождении.
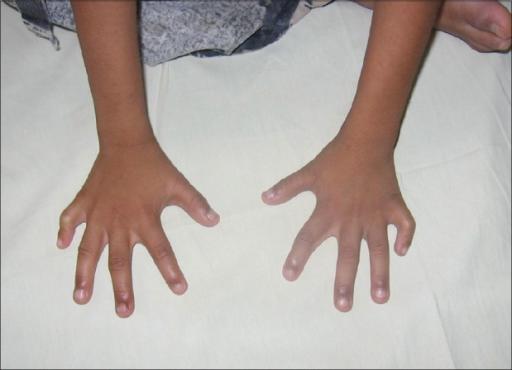
Хорошо заметны фенотипические признаки. Дети обладают низким лбом и маленьким ртом. Имеет место быть деформация ушных раковин и выступающий затылок. Присутствует расщелина нёба и верхней губы. Наблюдается птоз и деформация скелета. Присутствует характерная форма черепа. Иногда развивается аномалия ребер и врожденный вывих бедра. На ушах могут присутствовать козелки. Шея у ребенка короткая. Наблюдается косоглазие и косолапость. Почки имеют маленький размер. Наблюдаются скрещенные пальцы кистей. Стопа имеет форму качалки. На коже присутствуют папилломы и гемангиома.
Признаки синдрома Эдвардса
Уже через пару недель после рождения болезнь начинает себя проявлять. Симптомы, замеченные сразу, в дальнейшем остаются и могут усиливаться. Например, аномальный тонус постепенно переходит на все группы мышц, вызывая даже вывихи. На первом году жизни у ребенка обнаруживают заболевания важных систем органов.
Аномалии развития у новорожденных
Синдром Эдвардса сопровождают нарушения работы и отклонения практически во всех системах органов. У ребенка обнаруживаются такие аномалии:
- сниженный мышечный тонус;
- косоглазие;
- пренатальная гипотрофия (вес около 2200 г);
- асфиксии;
- суженные глазные щели;
- врожденные пороки сердца;
- крипторхизм у мальчиков;
- птоз (опущение верхнего века);
- ротовая щель небольших размеров;
- косолапость;
- укороченная грудина.
Внешние особенности детей с болезнью Эдвардса

За счет ярко выраженных внешних признаков трисомии 18 врачи могут заподозрить ее сразу после рождения. Это характерные изменения внешнего вида:
- Долихоцефалия – удлиненный и более узкий череп. В 80-85% случаев трисомии 18 этот признак выражен очень ярко. Чтобы увидеть диспропорцию, не нужно проводить специальные измерения.
- Микроцефалия – слишком маленький череп по отношению к туловищу. Встречается менее часто, чем долихоцефалия.
- Деформация и смещение ушной раковины. Как видно на фото, она располагается ниже, чем у здоровых людей. Выпуклости хряща отсутствуют, как и мочка с козелком. У 20-25% детей нет слухового канала, а у остальных он сильно сужен.
- Деформации неба. В месте, где должен быть срединный шов, проходит продольная щель. Возможно также незаращение мягкого и/или твердого неба, а иногда и губы.
- Стопа-качалка. Встречается у 75% больных. У ребенка выдается назад пяточный бугор, свода может не быть. Поверхность подошвы стопы плоская, а иногда даже выпуклая, что делает ее похожей на ножку кресла-качалки.
- Микрогнатия (микрогения) – слишком втянутый подбородок. У здоровых людей он выступает вперед. При небольших размерах головы 1 см между нижней и верхней челюстями считается слишком большим зазором, который ведет к подтеканию слюны. Симптом встречается у 70% детей с трисомией 18.
- Патологии развития наружных половых органов. У мальчиков недоразвит пенис, а у девочек гипертрофирован клитор. Такие аномалии не слишком распространены. Их обнаруживают у 15-20% детей с трисомией 18.
- Синдактилия – сращение пальцев, чаще на ногах. Его диагностируют в 45% случаев. При легких формах между пальцами просто образуется кожная складка, похожая на перепонку. В тяжелых – происходит сращение костной тканью.
- Флексорное положение кистей. Из-за постоянно напряженных сгибателей большой палец и мизинец прижаты к ладоням.
- Дерматоглифические признаки. Самым частым проявлением считается поперечная борозда, проходящая по всей ладони. Еще ее называют обезьяньей линией или линией Симиан. На подушечках пальцев можно заметить более частые дуги.
Проявления синдрома во взрослом возрасте
У врачей очень мало данных по поводу внешних признаков трисомии 18 в зрелом возрасте. Это связано с тем, что большая часть больных погибает еще в детском возрасте. Выживаемость несколько выше при мозаичной форме недуга, но и здесь случаи, когда ребенок дожил до совершеннолетия, единичны. Во взрослом возрасте болезнь проявляется так же. Характерные признаки:
- отсутствие эмоционального ответа на внешние раздражители;
- смещение ушной раковины и ее деформация;
- расщелины верхней губы и неба («заячья губа» или «волчья пасть»);
- умственная отсталость вплоть до имбецильности или идиотии;
- отсутствие реакции на резкие звуки.